
Maladie d’Huntington : un essai clinique porteur d’espoir
Un essai clinique en phase 1*, mené par une équipe de Londres, a permis pour la première fois de diminuer les quantités de la protéine toxique causant la maladie. Les résultats ont montré que ce traitement ne porte pas de risque et est bien toléré par les patients.
La maladie d’Huntington est une maladie neurodégénérative héréditaire. Les premiers symptômes apparaissent en général à l’âge adulte, entre 30 et 50 ans. Ils se manifestent par des troubles cognitifs et moteurs. A ce jour, les traitements existants agissent sur les symptômes mais ne permettent pas de ralentir la progression de la maladie.
Il s’agit d’une maladie génétique due à la mutation du gène contenant l’information nécessaire à la synthèse d’une protéine nommée huntingtine. La mutation du gène provoque l’expression d’une protéine anormale, qui va perturber le fonctionnement général du neurone et ainsi causer les symptômes de la maladie. Le gène est tout d’abord copié en une molécule » messager « , appelé ARN messager. Cet ARN messager est ensuite pris en charge par la cellule pour produire la protéine.
Le médicament testé lors de l’essai clinique, appelé Ionis-HTTRx, fonctionne en se fixant à la molécule messager et en provoquant ainsi sa destruction avant que la protéine toxique puisse être fabriquée. L’effet espéré était donc de réduire les quantités de protéines anormales dans le but de ralentir, voire de stopper la progression de la maladie. Des études sur modèles murins ont précédemment donné des résultats prometteurs. L’étape suivante lors de l’essai clinique de phase 1 consistait à s’assurer que ce traitement ne présente pas de risque chez l’humain.
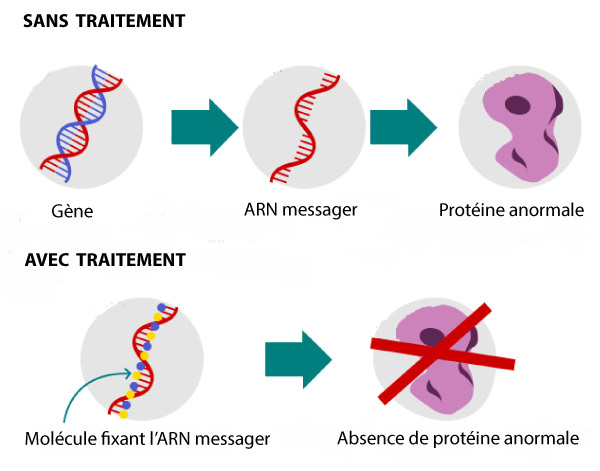
Après plusieurs années de développement pré-clinique, le premier essai clinique en phase 1 a démarré en 2015, mené par le professeur Sarah Tabrizi de l’UCL (University College of London). Il a été effectué sur 46 patients atteints d’une maladie d’Huntington en phase précoce. Pour que la molécule puisse atteindre le cerveau, le traitement a été administré par injection dans le liquide cérébro-spinal (liquide autour du cerveau et de la moelle épinière) au niveau du bas du dos. Les concentrations de protéines dans le liquide spinal ont été mesurées avant et après traitement. Comme espéré, le traitement diminue significativement les quantités de protéine mutée.
C’est une grande avancée car c’est la première fois qu’un médicament supprime, chez des patients, la présence de la protéine toxique causant la maladie. Les résultats positifs de cet essai clinique suggèrent que cette molécule expérimentale pourrait potentiellement ralentir la progression de la maladie. Un autre espoir serait de pouvoir adapter le traitement pour d’autres maladies du cerveau dues à une mutation qui est la cause bien définie de la maladie, en ciblant leur protéine anormale.
Un essai clinique plus large doit ensuite être réalisé pour tester si la molécule Ionis-HTTRx ralentit la progression de la maladie. En effet, l’essai clinique qui a été réalisé est un essai de phase 1, qui se déroule donc sur une courte durée et sur un faible nombre de patients. Les résultats exposés sont prometteurs mais ne permettent pas encore de connaitre avec certitude les effets sur la maladie. Des études sur modèles murins ont cependant montré qu’une diminution de la quantité de protéine anormale induit une réduction des symptômes de la maladie. Il faut maintenant étudier si cet effet est reproductible chez l’humain. Des essais de phase 2 et 3* doivent donc être effectués, avec de plus grandes cohortes et sur de plus longues durées. Ces étapes permettront de mesurer les effets précis du traitement sur les symptômes de la maladie d’Huntington.
En quoi consistent les essais cliniques ?
Évaluation de la toxicité de la molécule. Cela correspond à la première utilisation d’une nouvelle molécule chez l’humain. Le test est effectué durant de courtes périodes.
Mesure de l’efficacité de la molécule. Cette étape se déroule sur des durées plus longues. Elle permet d’étudier l’efficacité de la molécule et de déterminer la posologie adéquate.
Comparaison de l’efficacité de la molécule par rapport à un médicament déjà présent sur le marché ou à un placebo. Cette étape permet aussi de mesurer le rapport bénéfice/risque. Cette étape est réalisée sur un grand nombre de patients. C’est après validation de cette étape qu’un médicament peut être commercialisé.
Suivi après mise sur le marché du médicament. Cela permet entre autres d’étudier les effets secondaires indésirables et de faire un suivi à long terme.
Sources :
- FRC Neurodon : http://www.frcneurodon.org/informer-sur-la-recherche/actus/maladie-dhuntington-clinique-porteur-despoir/
- https://www.ucl.ac.uk/news/news-articles/1217/111217-huntingtons-disease-protein
- https://www.theguardian.com/science/2017/dec/11/excitement-as-huntingtons-drug-shown-to-slow-progress-of-devastating-disease
Retrouvez le portrait de Radhia Kacher, soutenue par la Fondation pour sa thèse sur la maladie d’Huntington
https://www.fondation-groupama.com/fiche/2015-radhia-kacher-maladie-de-huntington/